Boltz — A Next-Gen Open Source Engine for Biomolecular Modeling
What Is It?
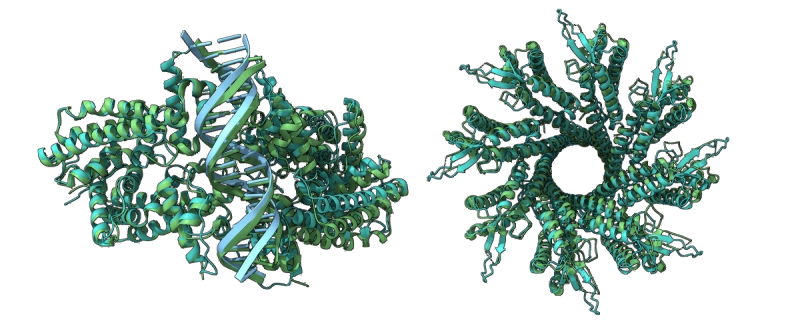
Boltz is an open-source family of deep learning models designed for predicting biomolecular interactions, including complex structures and binding affinities of macromolecules like proteins. Developed by Jeremy Wohlwend at MIT CSAIL, Boltz-1 is the first fully open-source model to match AlphaFold3-level accuracy in structural prediction, while Boltz-2 takes it further by unifying structure and affinity prediction. Remarkably, it achieves accuracy comparable to free energy methods at up to 1000× faster speeds.
Key Features
-
High-Precision Structure Prediction
Boltz‑1 delivers structural prediction for protein complexes on par with AlphaFold3, offering cutting-edge capabilities to the public. -
Fast and Accurate Affinity Estimation
Boltz‑2 rapidly predicts binding affinities between proteins and small molecules with accuracy comparable to FEP (Free Energy Perturbation) methods but with far lower compute requirements. -
Unified Structure + Affinity Modeling
Boltz‑2 integrates structure prediction and binding affinity estimation into a single model—ideal for tasks like virtual drug screening. -
Controlled Inference Capabilities
Users can guide predictions with custom structural constraints, such as binding pockets or MD (molecular dynamics) trajectories, for tailored results. -
Highly Optimized and Scalable
Boltz‑1x introduces CUDA acceleration and low-memory inference, making it suitable for both resource-limited and large-scale environments. -
Fully Open-Source (MIT License)
All code, weights, and utilities are freely available under the MIT license, supporting both academic and commercial use.
Technical Foundations
-
Diffusion + Transformer Architecture
Boltz builds on the Diffusion-Transformer design inspired by AlphaFold3, with enhancements in MSA processing, structure trimming, and pocket guidance. -
Confidence Estimation
Boltz‑1x includes AlphaFold3-style confidence heads that output pLDDT/pTM scores to estimate prediction reliability. -
Efficient Attention and Memory Management
Uses local atomic attention and chunked triangle attention to scale up to large biomolecular systems. -
Action-Conditioned Affinity Prediction
Boltz‑2 is trained via supervised learning on PubChem binding data to accurately model the influence of binding poses and structures on affinity.
Project Resources
Application Scenarios
-
Early-Stage Drug Discovery & Virtual Screening
Boltz‑2 can predict molecular binding structures and affinities in seconds, making large-scale virtual screening practically feasible. -
Academic Research in Complex Modeling
Boltz‑1 enables high-quality modeling of protein-protein, protein-RNA, and other molecular interactions for research labs. -
Molecular Optimization and Medicinal Chemistry
Affinity predictions allow chemists to evaluate how structural modifications impact binding, guiding compound optimization. -
Structure Conditioning & MD Integration
Supports user-defined input such as binding pockets or molecular dynamics trajectories for highly customized structure prediction. -
Community-Driven Open Science
With full open access to source code and weights, Boltz encourages collaborative development, research extensions, and novel applications in biomolecular modeling.
© Copyright Notice
The copyright of the article belongs to the author. Please do not reprint without permission.
Related Posts




No comments yet...